Overview of running a Phyre2 job |
|
|
Submitting a job |
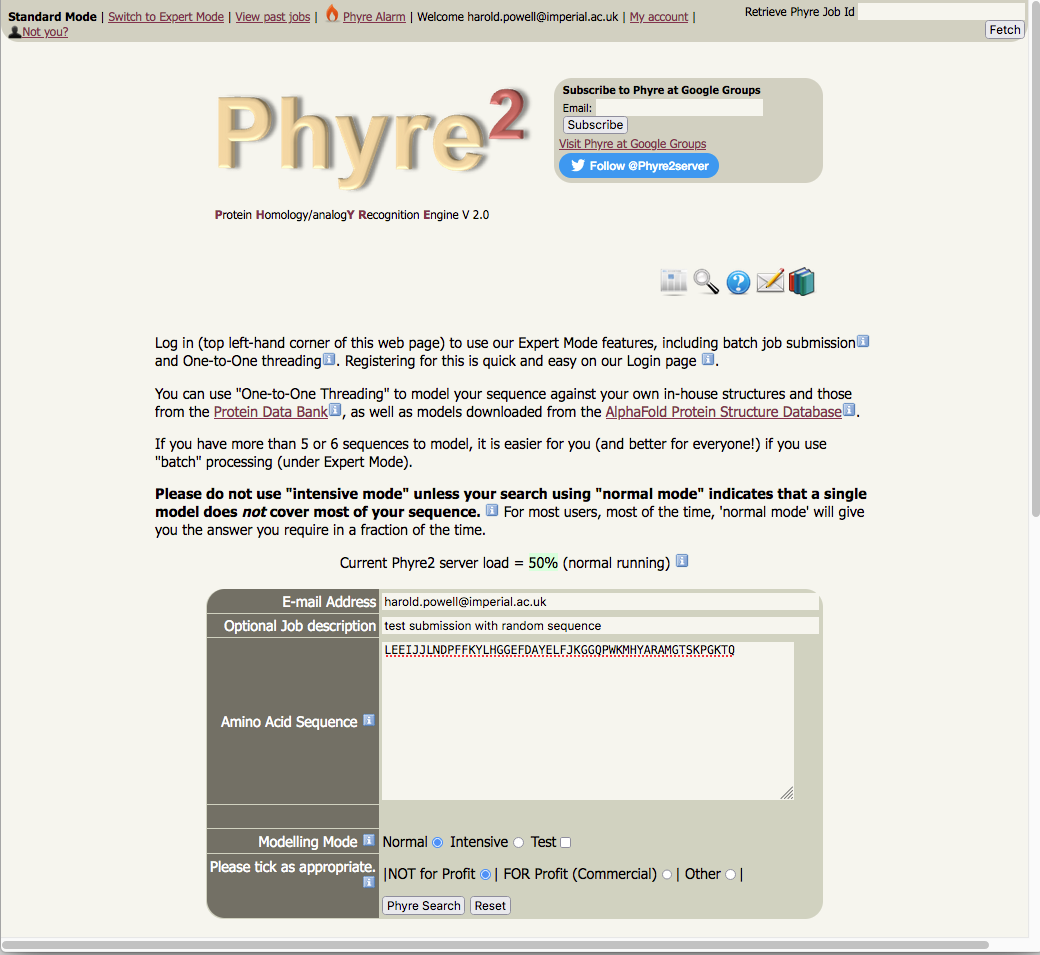
Running Phyre2 to obtain a model of an amino acid sequence is straightforward.
In most cases, all the user has to do is to enter
|
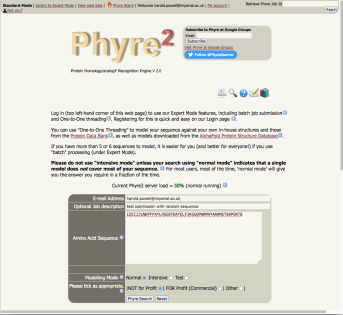
|
A note on Intensive mode and why you should not run it |
Intensive mode is
- not "better" than Normal mode except in one rare circumstance.
-
only of use (and even then it is somewhat limited and prone to error) if Normal mode shows that there are multiple models that cover different regions of your sequence. If there is a suitable single model that covers >~60 % of your sequence, the chances that Intensive mode will give a better model are small.
In a recent examination of results from a plant proteome (240,000 sequences), Phyre2
indicated that 48,000 might benefit from running intensive mode. Of these, ~4,000 would
have given a model more than about 40 residues longer than normal mode - so it could be worth
running intensive mode for about 1 in 60 sequences.
-
very slow compared to Normal mode; it typically takes 4 - 10 times as long to run a job, and in nearly every case gives the same final answer.
|
A note on Intensive mode and
when you should run it |
|
If the results from normal mode state explicitly that it would be worth running
intensive mode, then that is the time to run it. Apart
from that specific case, you are probably wasting your
time if you do, because normal mode will give you the
answer you really want.
See
|
After submitting a job |
|
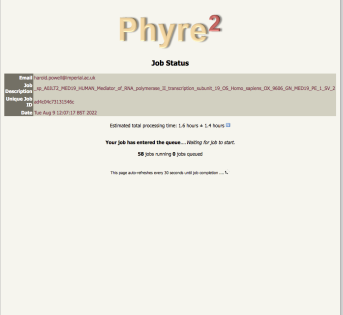
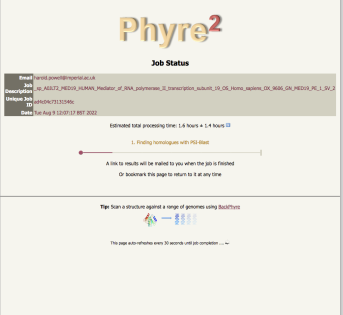
The interface changes and gives you information on the progress of your job;
|
|
|
|
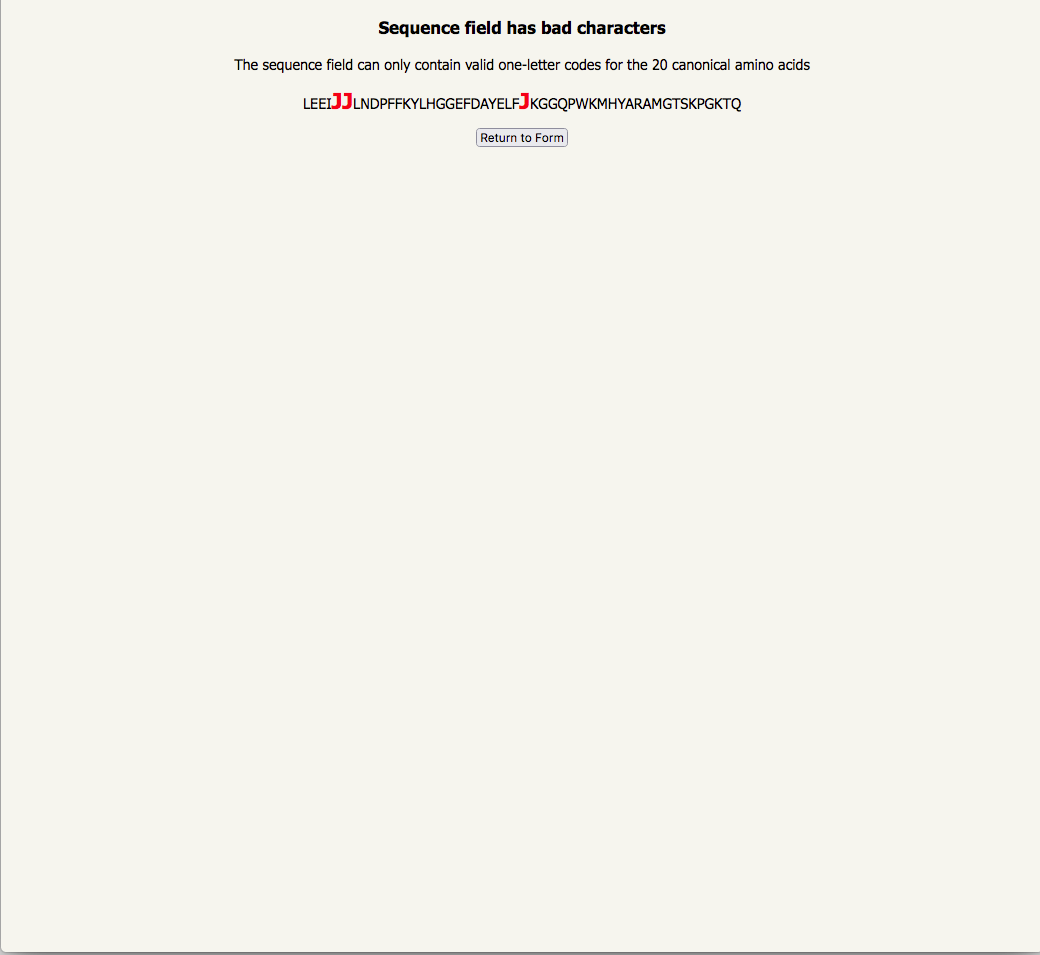
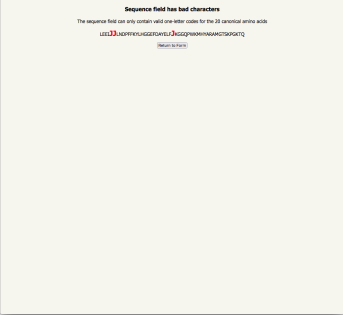
| if you have entered a non-canonical amino acid, the unacceptable residues are highlighted |
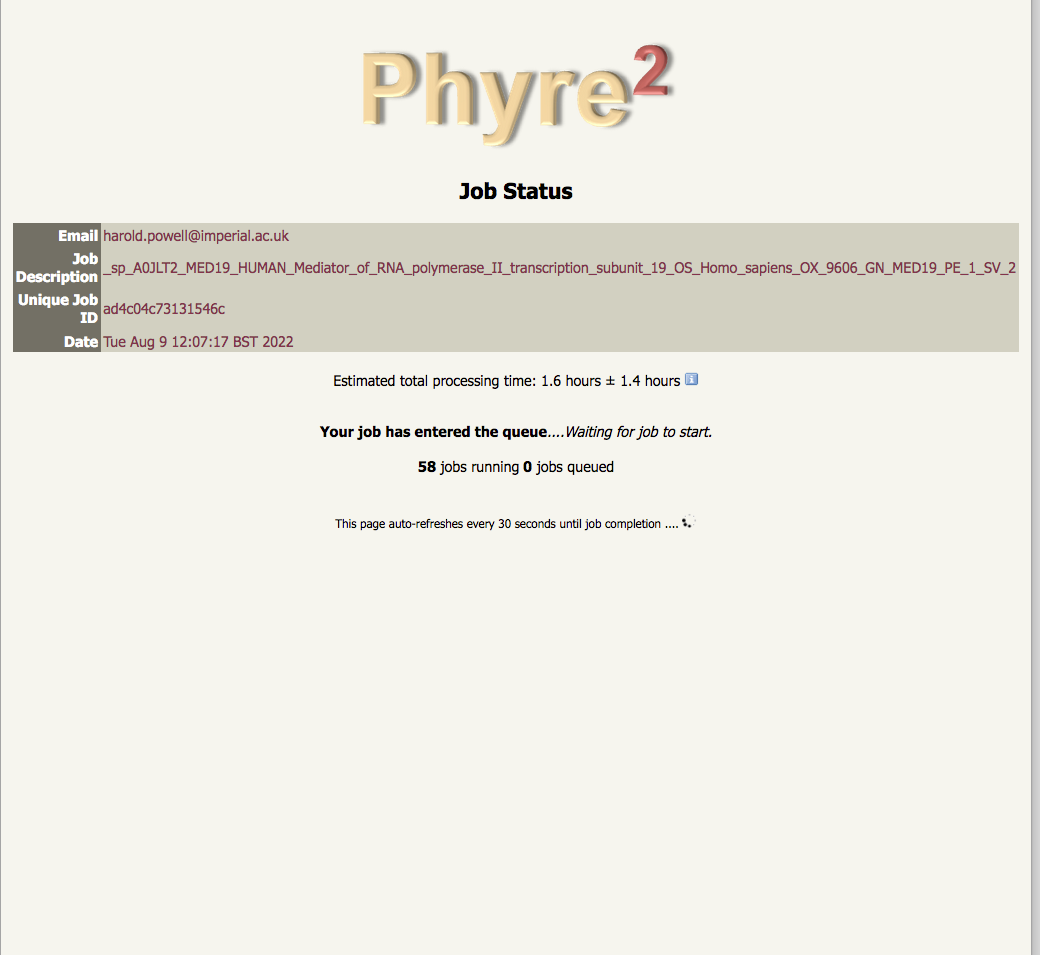
for normal jobs we give a very rough estimate of how long it might take, but for intensive jobs we don't |
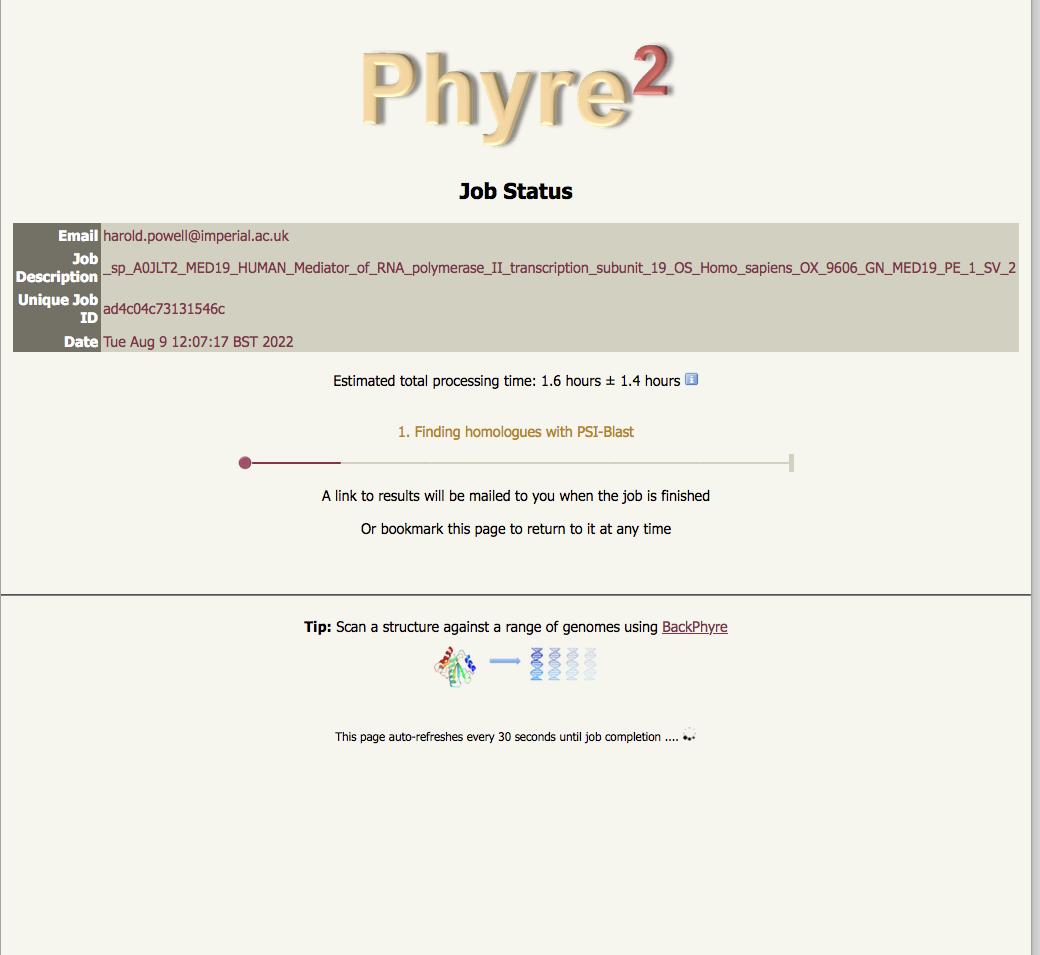
progress report - there are 5 major steps |
On completion |
|
If you have stayed on the page, it will automatically update to the results page when the job
has finished. Otherwise, you can follow the link in the results e-mail:
|
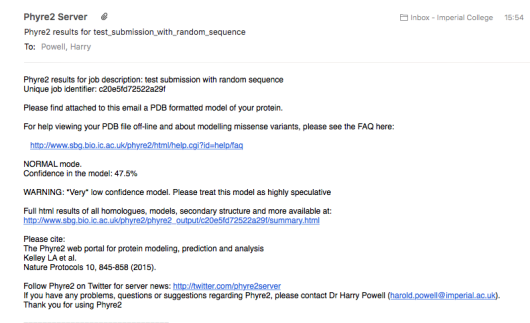
|
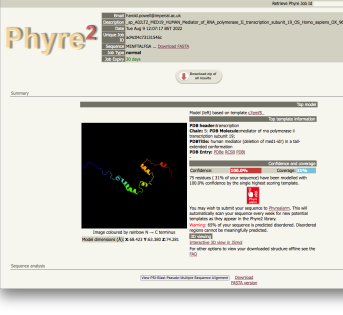
Results |
|
|
|
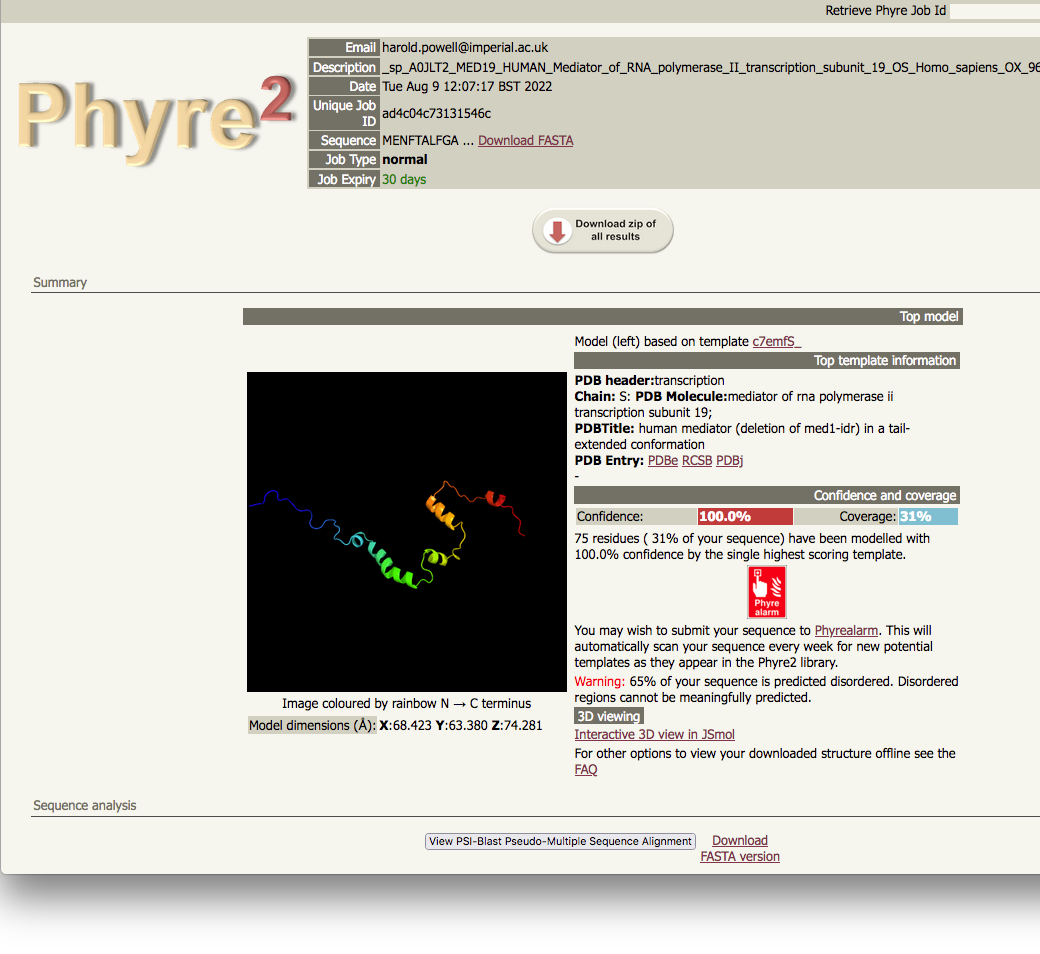
| normal job results |
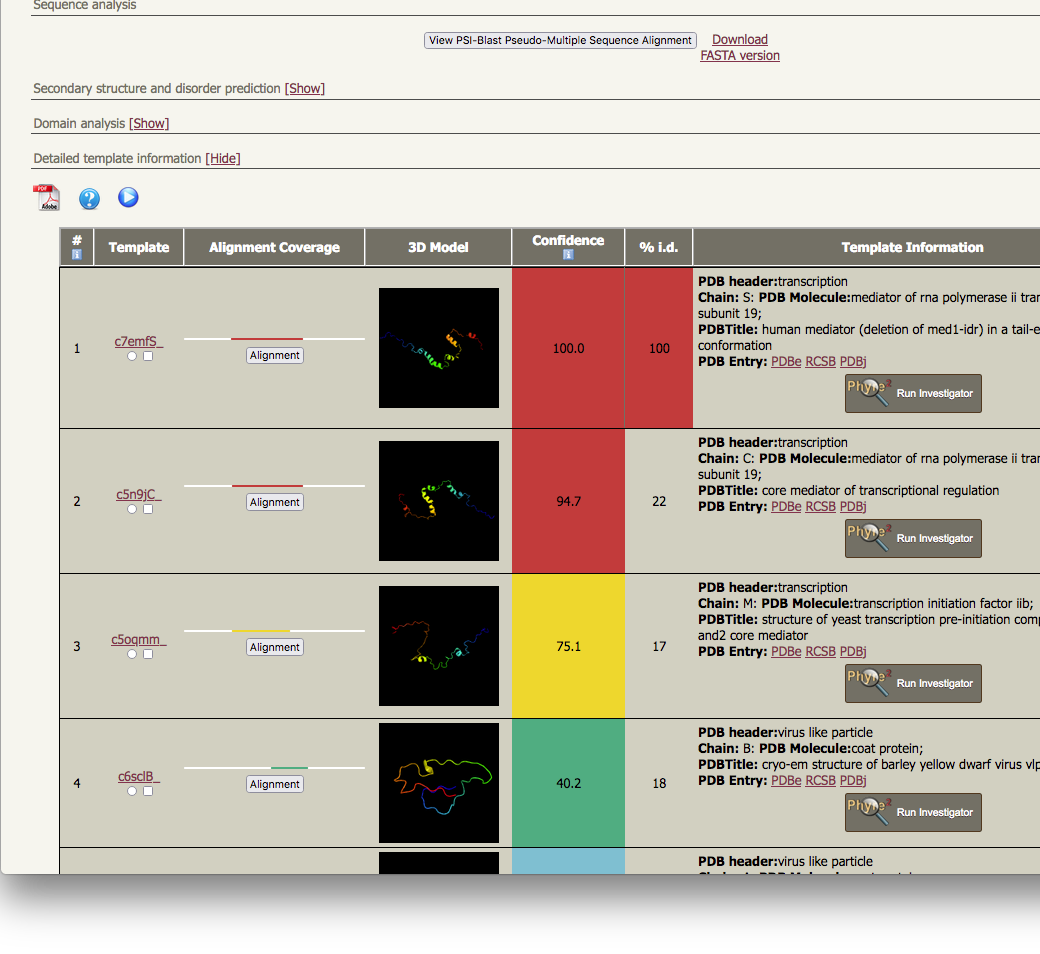
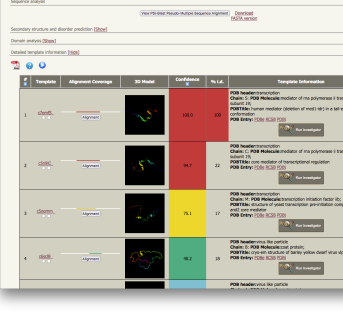
the top 20 hidden Markov Models are all modelled by Phyre2 |
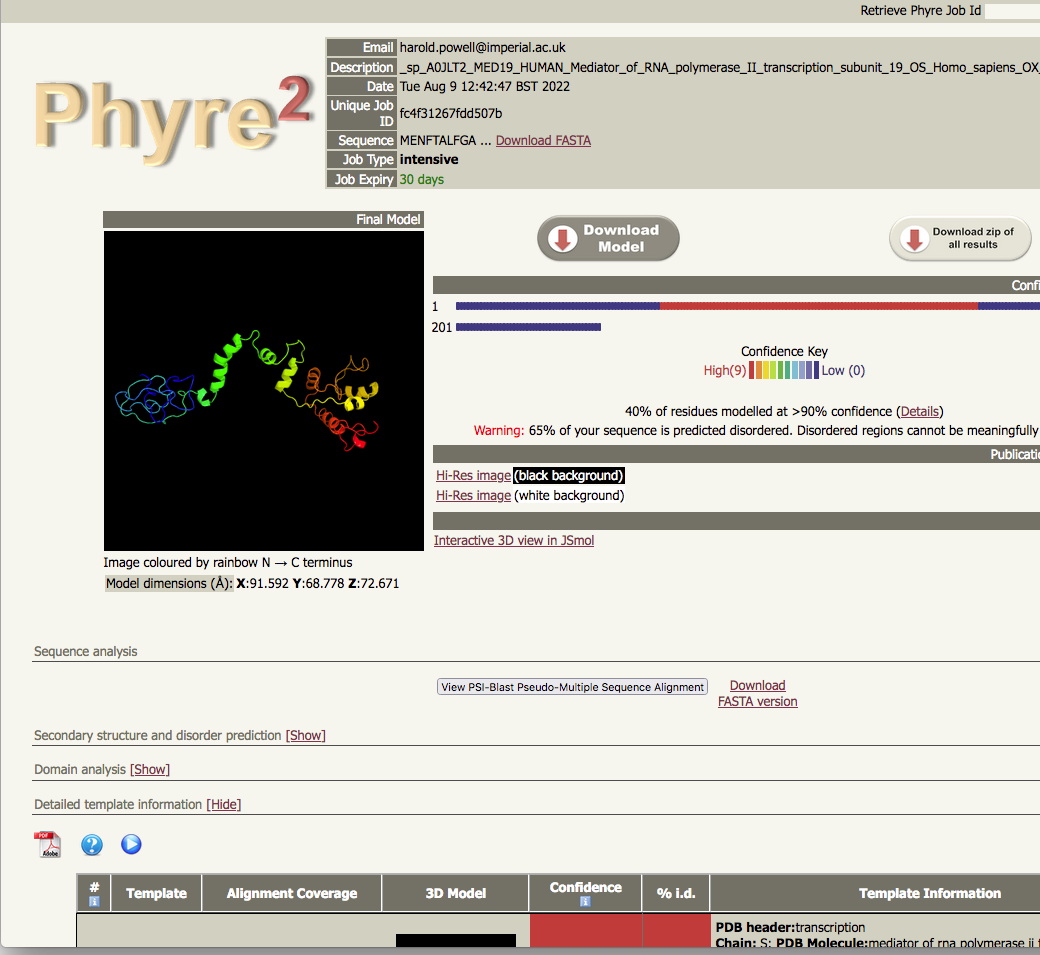
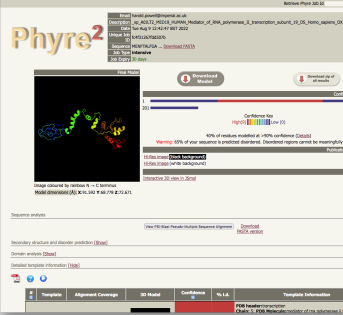
intensive job results |