|
| |||||
| Protein Homology/analogY Recognition Engine V 2.0 | ||||||
|
|
|
| |||||
| Protein Homology/analogY Recognition Engine V 2.0 | ||||||
|
Interpreting Results: Intensive |
1. Overview/pipeline'Intensive' modelling by Phyre2 is identical to the 'normal' mode up to the point of model construction. All the same results can be found in an intensive run as in the normal run. However, once models have been constructed Phyre2:
Please Note: Intensive mode is currently limited to modelling sequences < 1000 residues in length. At the top of the results page for an intensive job is a summary like that shown below: |
|
|
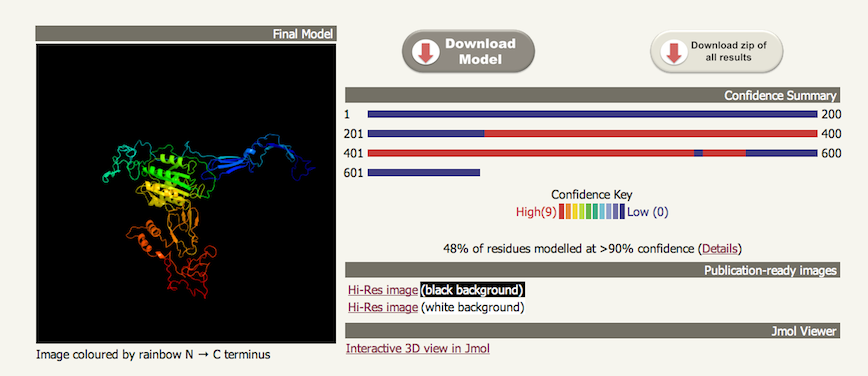
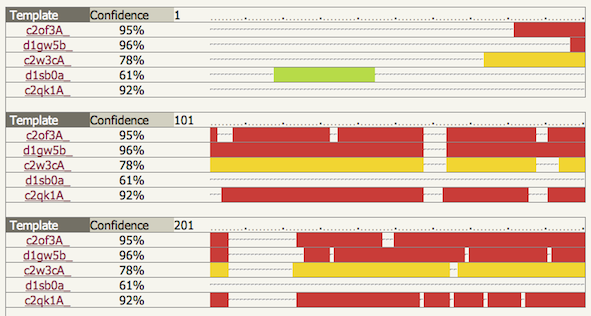
An image of the model of the complete protein is shown on the left. On the right is a summary figure to illustrate the confidence in the model along its length. Confidence is coloured from high (red) to low (blue) using a rainbow spectrum. Note: when multiple independent domains are modelled without a template that covers all these domains, the relative domain orientation is highly unreliable. A summary statistic is reported below the graphic showing what percentage of the residues have been modelled at >90% confidence. For confidence please see the 'Interpreting results: normal' . Next to this statistic is a "Details" link (see below). Links are also present to high resolution ray traced images of the model. Finally the link at the bottom of this section launches the JMol java applet within the page for interactive 3D views of the model. It is also possible within this view to colour the structure by confidence so you can assess which parts of the model in 3D space are considered trustworthy. 2. Multi-template detailsIf you click on the 'Details' link next to the summary statistic it will take you to a table at the bottom of the results page. This table shows which templates were used as a source of constraints coloured by confidence. The regions covered by these templates are shown together with the confidence in the match. An example can be seen below: |
|
|
|
In this example 5 templates have been used, 2 of which have borderline confidence of 61% and 78%. Regions (or rather columns) where no template is present have been modelled by the ab initio component of the Poing system. ab initio protein modelling is notoriously hard and most certainly an unsolved problem. Note: consider regions that have modelled by ab initio with *extreme* caution. Hopefully they may provide you with some hints, but do not trust them to be correct. |
Phyre is now FREE for commercial users!
All images and data generated by Phyre2 are free to use in any publication with acknowledgement
Accessibility Statement| Please cite: The Phyre2 web portal for protein modeling, prediction and analysis | |||||||||
| Kelley LA et al. Nature Protocols 10, 845-858 (2015) [paper] [Citation link] | |||||||||
|
| ||||||||