



The pie-charts were generated similar to those measuring the extend of structure assignment for the two genomes. Coiled-coil and transmembrane regions are not shown in separate fractions of the pie-chart because these can be matched by sequences of our database (i.g. most of these regions are in fact matched by sequence homologues, data not shown). Although our benchmark is based on protein structure it is mainly PSI-BLAST that determines the success of finding a homologue for a given query sequence and we transfer the results of the benchmark to sequence pure information. The ration of undetected remote homologies to detected remote homologies as determined by our benchmark) (2.1) is used to estimate the fraction of undetected homologues in the two genomes.
The results for MG show that the information for nearly complete annotation of the genome is potentially in the public sequence databases. The remaining one percent of the genome may there represent the small bit of new information, e.g. an MG specific pathway or a small collection of parasite/host factors. The MG genome is the smallest fully sequenced bacterial genome currently available (479 genes), and is may be not more than the minimal set of genes required for cellular life ( Mushegian AR, Koonin EV (1998) Proc Natl Acad Sci U S A 93, 10268-73 ). Compared to MG the genome of TB a lot more secrets in it's genome.
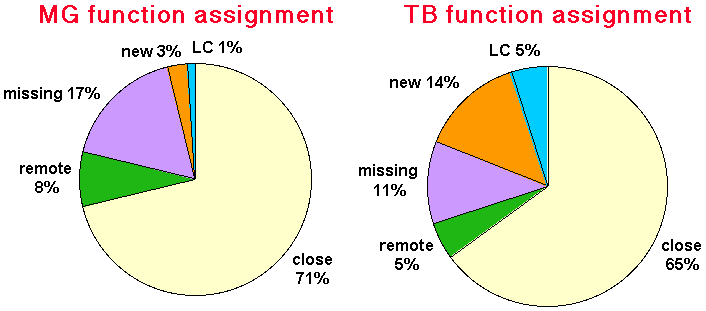
Legend: LC (low complexety regions), close (machtes by close homologues), remote (matches by remote homologues only), missing (estimated undetected remote homologies) new superfams (fraction of genes with potentially new function, i.g. found not in any sequence and structure database)
get functional annotations ...

Copyright © 1999-2002 Cancer Research UK
All Rights Reserved, disclaimer
Comments to author: a.mueller@cancer.org.uk
Generated: Thu Jun 27, 2002