



The result of our benchmark are shown below. In the table sequences are chains (single or multi-domain proteins). For sequences 'correctly assigned' means there is at least one domain correctly assigned regardless whether there are also false assignments in the same sequence. Accordingly 'false' in sequences context means there is at least one false domain assignment in the sequence. An 'over assignment' is e.g. when two domains are assigned to a region in the query that represents only one domain. A 'Correct' assignment is when the domain of the query and the assignment (target) are of the same superfamily. 'Residues' is the sum of domain length.
| Description | Sequences | Domains | Residues |
|---|---|---|---|
| No. in SCOP genome | 1254 | 1621 | 263,863 |
| No. correctly assigned | 503 | 652 | 84,827 |
| Coverage of correct assignments(%) | 40 | 40 | 32 |
| No. of false positive assignments | 13 | 16 | 1,985 |
| No. of over assignments | 2 | 2 | 163 |
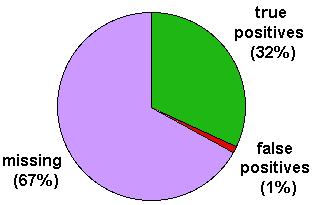
The pie-chart summarises the results (bases on residues). 'missing' are assignments that could have been found by PSI-BLAST (because there are homologues proteins in the target database). The ration of missing to true positives is 2.1 and can be used to estimate the fraction of undetected remote homologues in real genomes.

Copyright © 1999-2002 Cancer Research UK
All Rights Reserved, disclaimer
Comments to author: a.mueller@cancer.org.uk
Generated: Thu Jun 27, 2002