| Secondary structure and disorder prediction | |
 |
| | |
1 | . | . | . | . | . | . | . | . | 10 | . | . | . | . | . | . | . | . | . | 20 | . | . | . | . | . | . | . | . | . | 30 | . | . | . | . | . | . | . | . | . | 40 | . | . | . | . | . | . | . | . | . | 50 | . | . | . | . | . | . | . | . | . | 60 |
| Sequence | |
M | I | Y | L | V | I | S | V | F | L | I | T | A | F | I | C | L | Y | L | K | K | D | I | F | Y | P | A | V | C | V | N | I | I | F | A | L | V | L | L | G | Y | E | I | T | S | D | I | Y | A | F | Q | L | N | D | A | T | L | I | F | L |
| Secondary structure | |
|  | | | | | | | | | | | | | | | | | | | |
|
|
|
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| SS confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder | |
? | ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| | |
. | . | . | . | . | . | . | . | . | 70 | . | . | . | . | . | . | . | . | . | 80 | . | . | . | . | . | . | . | . | . | 90 | . | . | . | . | . | . | . | . | . | 100 | . | . | . | . | . | . | . | . | . | 110 | . | . | . | . | . | . | . | . | . | 120 |
| Sequence | |
L | C | N | V | L | T | F | T | L | S | C | L | L | T | E | S | V | L | D | L | N | I | R | K | V | N | N | A | I | Y | S | I | P | S | K | K | V | H | N | V | G | L | L | V | I | S | F | S | M | I | Y | I | C | M | R | L | S | N | Y | Q |
| Secondary structure | |
| | | | | | | | | | | | | | | | |
|
|
|
|
|
|
|
|
|
|
|  | |  |
|
|
|
|
|
|
|
| | | | | | | | | | | | | | | | | | | | | |
| SS confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? |
| ? | ? | ? | ? | ? | ? | ? |
| ? | ? | ? |
|
|
|
|
| ? | ? | ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| | |
. | . | . | . | . | . | . | . | . | 130 | . | . | . | . | . | . | . | . | . | 140 | . | . | . | . | . | . | . | . | . | 150 | . | . | . | . | . | . | . | . | . | 160 | . | . | . | . | . | . | . | . | . | 170 | . | . | . | . | . | . | . | . | . | 180 |
| Sequence | |
F | G | T | S | L | L | S | Y | M | N | L | I | R | D | A | D | V | E | D | T | S | R | N | F | S | A | Y | M | Q | P | I | I | L | T | T | F | A | L | F | I | W | S | K | K | F | T | N | T | K | V | S | K | T | F | T | L | L | V | F | I |
| Secondary structure | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| SS confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? | ? | ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| | |
. | . | . | . | . | . | . | . | . | 190 | . | . | . | . | . | . | . | . | . | 200 | . | . | . | . | . | . | . | . | . | 210 | . | . | . | . | . | . | . | . | . | 220 | . | . | . | . | . | . | . | . | . | 230 | . | . | . | . | . | . | . | . | . | 240 |
| Sequence | |
V | F | I | F | A | I | I | L | N | T | G | K | Q | I | V | F | M | V | I | I | S | Y | A | F | I | V | G | V | N | R | V | K | H | Y | V | Y | L | I | T | A | V | G | V | L | F | S | L | Y | M | L | F | L | R | G | L | P | G | G | M | A |
| Secondary structure | |
| | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
|
|
|
|
| | | |
| SS confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? | ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? |
| ? | ? | ? |
|
| ? |
|
|
| Disorder confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| | |
. | . | . | . | . | . | . | . | . | 250 | . | . | . | . | . | . | . | . | . | 260 | . | . | . | . | . | . | . | . | . | 270 | . | . | . | . | . | . | . | . | . | 280 | . | . | . | . | . | . | . | . | . | 290 | . | . | . | . | . | . | . | . | . | 300 |
| Sequence | |
Y | Y | L | S | M | Y | L | V | S | P | I | I | A | F | Q | E | F | Y | F | Q | Q | V | S | N | S | A | S | S | H | V | F | W | F | F | E | R | L | M | G | L | L | T | G | G | V | S | M | S | L | H | K | E | F | V | W | V | G | L | P | T |
| Secondary structure | |
| | | | | | | | | | | | | | | | | |
|
|
|
|
| | | | | | | | | | | | | | | | | | |
|
|
|
|
| | | | | | | |
|
|
|
|
|
|
|
| SS confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? | ? | ? |
| ? |
| ? | ? |
|
|
|
| ? |
| ? |
|
|
|
|
|
| Disorder confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| | |
. | . | . | . | . | . | . | . | . | 310 | . | . | . | . | . | . | . | . | . | 320 | . | . | . | . | . | . | . | . | . | 330 | . | . | . | . | . | . | . | . | . | 340 | . | . | . | . | . | . | . | . | . | 350 | . | . | . | . | . | . | . | . | . | 360 |
| Sequence | |
N | V | Y | T | A | F | S | D | Y | V | Y | I | S | A | E | L | S | Y | L | M | M | V | I | H | G | C | I | S | G | V | L | W | R | L | S | R | N | Y | I | S | V | K | I | F | Y | S | Y | F | I | Y | T | F | S | F | I | F | Y | H | E | S |
| Secondary structure | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
|
|
| | | | | | | | | | | | | | | | | | | | | |
| SS confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| | |
. | . | . | . | . | . | . | . | . | 370 | . | . | . | . | . | . | . | . | . | 380 | . | . | . | . | . | . | . | . |
| Sequence | |
F | M | T | N | I | S | S | W | I | Q | I | T | L | C | I | I | V | F | S | Q | F | L | K | A | Q | K | I | K |
| Secondary structure | |
| | | | | | | | | | | | | | | | | | | | | |
|
|
|
|
|
|
| SS confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? | ? | ? | ? | ? | ? |
| Disorder confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| Confidence Key |
| High(9) | |
|
|
|
|
|
|
|
|
|
Low (0) |
| ? | Disordered |
| Alpha helix |
| Beta strand |
Hover over an aligned region to see model and summary info
Please note, only up to the top 20 hits are modelled to reduce computer load
|
| 1 |
|
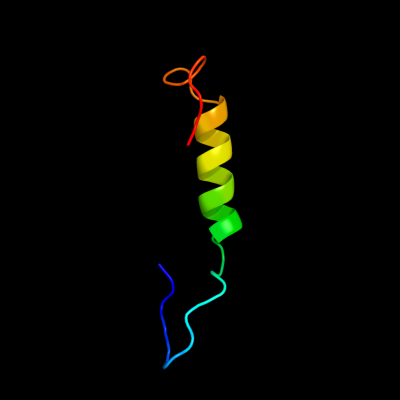
PDB 1ohe chain A domain 1
Region: 305 - 342
Aligned: 38
Modelled: 38
Confidence: 6.4%
Identity: 8%
Fold: (Phosphotyrosine protein) phosphatases II
Superfamily: (Phosphotyrosine protein) phosphatases II
Family: Dual specificity phosphatase-like
Phyre2
| 2 |
|
PDB 2hac chain A
Region: 320 - 336
Aligned: 17
Modelled: 17
Confidence: 6.2%
Identity: 24%
PDB header:membrane protein
Chain: A: PDB Molecule:t-cell surface glycoprotein cd3 zeta chain;
PDBTitle: structure of zeta-zeta transmembrane dimer
Phyre2
|
| Detailed template information | |
Due to computational demand, binding site predictions are not run for batch jobs
If you want to predict binding sites, please manually submit your model of choice to 3DLigandSite
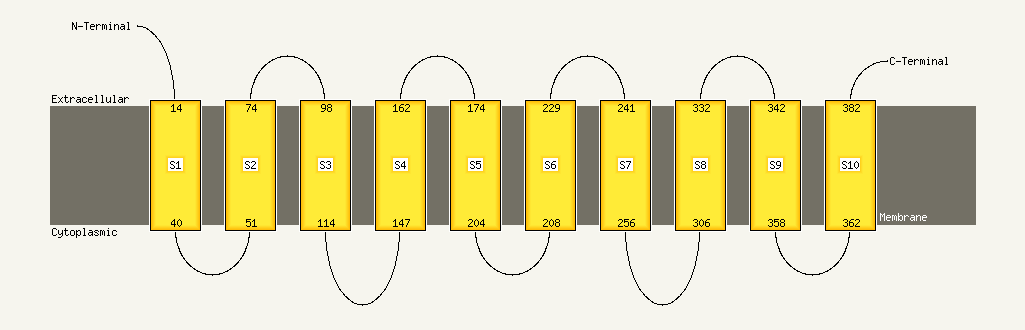
| Transmembrane helix prediction | |
Transmembrane helices have been predicted in your sequence to adopt the topology shown below
Phyre is for academic use only
| Please cite: Protein structure prediction on
the web: a case study using the Phyre server |
| Kelley LA and Sternberg MJE. Nature Protocols
4, 363 - 371 (2009) [pdf] [Import into BibTeX] |
| |
| If you use the binding site
predictions from 3DLigandSite, please also cite: |
| 3DLigandSite: predicting ligand-binding sites using similar structures. |
| Wass MN, Kelley LA and Sternberg
MJ Nucleic Acids Research 38, W469-73 (2010) [PubMed] |
| |
|
|
|
|