| Secondary structure and disorder prediction | |
| | |
1 | . | . | . | . | . | . | . | . | 10 | . | . | . | . | . | . | . | . | . | 20 | . | . | . | . | . | . | . | . | . | 30 | . | . | . | . | . | . | . | . | . | 40 | . | . | . | . | . | . |
| Sequence | |
M | M | F | E | F | N | M | A | E | L | L | R | H | R | W | G | R | L | R | L | Y | R | F | P | G | S | V | L | T | D | Y | R | I | L | K | N | Y | A | K | T | L | T | G | A | G | V |
| Secondary structure | |
|
|  |  |
|
|  | | | | | | | | | |
| | | | | | |
| | | | | | | | | | | | | | | | | | |
|
|
|
|
| SS confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Disorder | |
? | ? | ? | ? | ? | ? |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ? | ? | ? | ? | ? | ? |
| Disorder confidence | |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
| Confidence Key |
| High(9) | |
|
|
|
|
|
|
|
|
|
Low (0) |
| ? | Disordered |
| Alpha helix |
| Beta strand |
Hover over an aligned region to see model and summary info
Please note, only up to the top 20 hits are modelled to reduce computer load
|
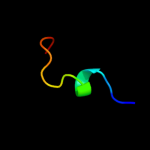
| 1 |
|
PDB 3b8f chain B
Region: 4 - 17
Aligned: 14
Modelled: 14
Confidence: 6.9%
Identity: 21%
PDB header:hydrolase
Chain: B: PDB Molecule:putative blasticidin s deaminase;
PDBTitle: crystal structure of the cytidine deaminase from bacillus anthracis
Phyre2
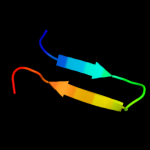
| 2 |
|
PDB 1rq0 chain A
Region: 17 - 33
Aligned: 17
Modelled: 17
Confidence: 6.1%
Identity: 47%
Fold: Release factor
Superfamily: Release factor
Family: Release factor
Phyre2
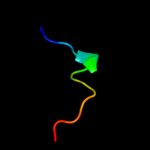
| 3 |
|
PDB 1taf chain A
Region: 21 - 39
Aligned: 19
Modelled: 19
Confidence: 5.8%
Identity: 32%
Fold: Histone-fold
Superfamily: Histone-fold
Family: TBP-associated factors, TAFs
Phyre2
| 4 |
|
PDB 3eud chain E
Region: 18 - 30
Aligned: 12
Modelled: 13
Confidence: 5.6%
Identity: 58%
PDB header:nuclear protein
Chain: E: PDB Molecule:protein shq1;
PDBTitle: structure of the cs domain of the essential h/aca rnp2 assembly protein shq1p
Phyre2
|
| Detailed template information | |
Due to computational demand, binding site predictions are not run for batch jobs
If you want to predict binding sites, please manually submit your model of choice to 3DLigandSite
Phyre is for academic use only
| Please cite: Protein structure prediction on
the web: a case study using the Phyre server |
| Kelley LA and Sternberg MJE. Nature Protocols
4, 363 - 371 (2009) [pdf] [Import into BibTeX] |
| |
| If you use the binding site
predictions from 3DLigandSite, please also cite: |
| 3DLigandSite: predicting ligand-binding sites using similar structures. |
| Wass MN, Kelley LA and Sternberg
MJ Nucleic Acids Research 38, W469-73 (2010) [PubMed] |
| |
|
|
|
|