



Next: Browse the results
Up: Results
Previous: Parameters setup
In constructing rules, Progol looks for motifs which are common to all
the domains of a given fold but almost never encountered in others,
except for a limited number cases which is set by a user defined
threshold (noise). Features which are important for structure and/or
function tend to be conserved amongst members of the same fold, at
least up to the level of super-family. Hence the rules learnt by
Progol should be useful in identifying those conserved motifs. In
this section we review six rules which were produced automatically
and present a possible biological interpretation. The complete set of
rules is available on our Web site.
Rule 1 (Globin fold)
Helix A at position 1 is followed by helix B. B contains a
proline residue.
The Globin-fold is good example of divergent evolution. In
SCOP, this fold comprises diverse sequences such as
myoglobin, hemoglobin and phycocyanins (but not colicins). Yet the
three-dimensional structure of these proteins is well preserved. One
hallmark of this fold is presence of a conserved proline residue in
helix B, which causes a sharp bend in the main chain. This
observation has been reported previously by Bashford et
al. [7], see the corresponding
Web page.
Rule 2 (4-helical cytokines)
The first helix is long and followed by another
helix.
Rule 3 (4-helical cytokines)
The second strand is immediately followed by a
helix.
Often, Progol produces more than one rule to cover all the positive
examples of a fold. Similarly, SCOP classification has more
than one family and/or more than one super-family per fold. Thus,
sometimes the mapping of the rules onto the examples matches that of
SCOP. This occurs for the 4-helical cytokines, which has two
families, the long-chain and short-chain cytokines. Members of the
long-chain cytokines family all start with a long helix, as observed
by Progol, see Rule 2. While the distinctive feature
for the short-chain cytokines is the absence of a coil between the
last strand-helix pair, Rule 3. Although these
proteins have been classified in the same family, their sequences are
quite diverged (with pairwise distances within the so-called twilight
zone).
Rule 4 (lambda repressor)
Helix C at position 3 is followed by helix D. The protein is between
53 and 88 residues long. The coil between C and D is about 6 residues
long.
Cro and repressor, of all three bacteriophages, 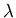 ,
434 and
P22, bind DNA in a similar way. The second helix of a helix-turn-helix
motif makes sequence-specific contacts with the edge of base pairs
situated in the major groove of the DNA molecule,
see the corresponding Web page. The specificity for the different
operator regions is the result of a surface complementarity. Specific
DNA sequence, TA-rich region, allows for specific deformation which
makes favorable contacts with the loop region between the second helix
of the helix-turn-helix motif and the following one. Which covers the following
bacteriophage domains: lambda C1 repressor (1lmb), 434 C1 repressor
(2r63), cro 434 (2cro) and P22 C2 repressor (1adr), but also in the
eukaryotic Oct-1 POU-specific domain (1pou). The POU-specific domain
contains a helix-turn-helix motif and binds DNA in a similar way to
the prokaryotic transcription factors Cro and repressor. Based on
this evidence a common origin has been suggested. Here, we
find that the similarity also extents to the conservation of the
length of the loop between the recognition helix (C) and the one that
follows (D).
,
434 and
P22, bind DNA in a similar way. The second helix of a helix-turn-helix
motif makes sequence-specific contacts with the edge of base pairs
situated in the major groove of the DNA molecule,
see the corresponding Web page. The specificity for the different
operator regions is the result of a surface complementarity. Specific
DNA sequence, TA-rich region, allows for specific deformation which
makes favorable contacts with the loop region between the second helix
of the helix-turn-helix motif and the following one. Which covers the following
bacteriophage domains: lambda C1 repressor (1lmb), 434 C1 repressor
(2r63), cro 434 (2cro) and P22 C2 repressor (1adr), but also in the
eukaryotic Oct-1 POU-specific domain (1pou). The POU-specific domain
contains a helix-turn-helix motif and binds DNA in a similar way to
the prokaryotic transcription factors Cro and repressor. Based on
this evidence a common origin has been suggested. Here, we
find that the similarity also extents to the conservation of the
length of the loop between the recognition helix (C) and the one that
follows (D).
Rule 5 (Rossmann fold)
Strand A at position 1 is followed by helix B. Strand C at position 6
is followed by helix D. The coil between A and B is about one residue
long.
NAD-binding domains of the Rossmann fold all have a similar binding
mechanism. The adenosine is bound to the short loop between the first
strand and the following helix. The region is embedded in a
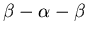 motif which has a highly conserved and contains
the sequence motif G-X-G-X-X-G. The fifth and sixth
secondary structures clamp the nicotinamide moiety of NAD,
see the corresponding Web page.
motif which has a highly conserved and contains
the sequence motif G-X-G-X-X-G. The fifth and sixth
secondary structures clamp the nicotinamide moiety of NAD,
see the corresponding Web page.
Rule 6 (P-loop)
Strand A at position 1 is followed by helix B. The coil between A and
B is about 5 residues long.
The first loop of the P-loop fold is necessary for the proper binding
of the guanine nucleotide, see the corresponding Web page. The loop is
also called diphosphate-binding loop or P-loop, and gives its name to
the fold. The strand-loop-helix region contains the conserved
sequence motif, G-X-X-X-X-G-K-S/T, which is used in PROSITE
database [8] to characterise the ATP/GTP-binding motif.
Next: Browse the results
Up: Results
Previous: Parameters setup
Marcel Turcotte
1999-10-20