Phyrestorm
Phyre Searching TOpology by Rapid Matching
| Home | About | Help | Contact |
Help
| Introduction |
| PhyreStorm first searches a clustered form of the Protein Data Bank (PDB). Each structural cluster is represented by a single protein called the medoid (the structure closest to the centre of the cluster). If such a medoid matches your protein sufficiently well (above the threshold set at submission time), further searches will be performed on the remaining members of the cluster. The initial results screen shows the cluster representatives matched by your protein. |
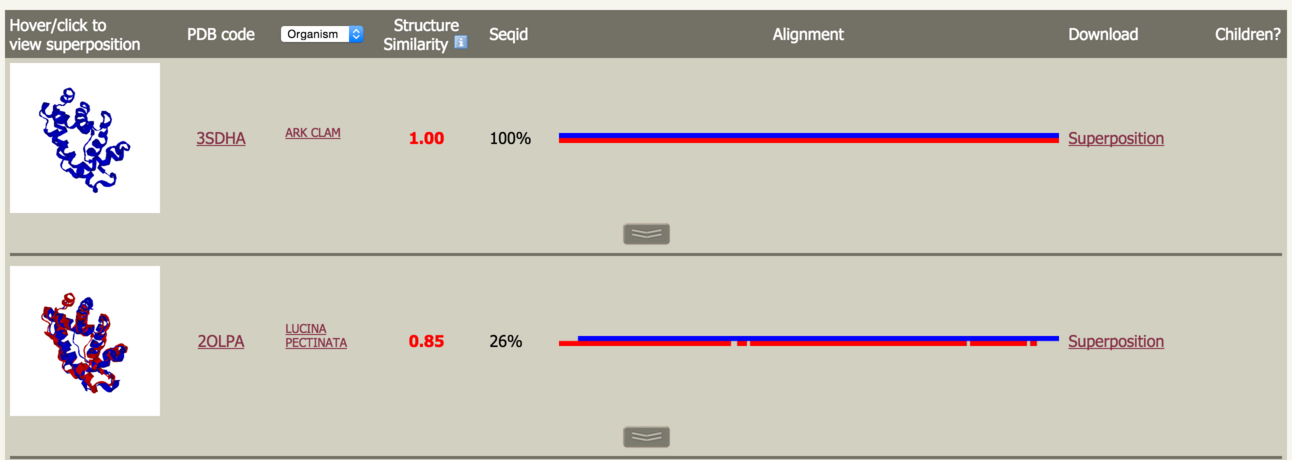 |
Hover over the protein image to pop-up a window containing a static image of the superposition. Click to launch the 3DMol interactive molecular viewer to inspect the superposition. Close this window by clicking the X in the top right. |
Throughout PhyreStorm, you can differentiate your protein from the one matched using the colour key at the top of the page: |
 |
Click on the PDB code to be taken to the RCSB. Next to the PDB code header is a drop down list to allow you to select one of several different types of PDB information to display for the matches: Organism, PDB title and Keywords. Next is the Structure Similarity score as measured by the TMscore. This score ranges from 0-1 where 1 is a perfect match and >0.5 indicates, usually, the same protein fold. Following this is the sequence identity between your protein and the matched protein. It is particularly interesting to note cases where the structures are highy similar at extremely low sequence identities. This illustrates cases where structural alignment is able to uncover relationships otherwise undetectable by sequence searching. |
Alignment |
 |
This is an image of the alignment generated by TMalign. The colours correspond to the key at the top of the page. |
| This image allows you at a glance to assess the degree of 'gappiness' of the alignment and to what extent one protein may have matched a substructure of the other. Clicking on this image will pop-up a window as shown below: |
 |
| Green indicates closely aligned residues, while yellow indicates greater deviation in the superposition. Click the appropriate link to download the alignment in the format you require. |
Superposition |
| This is a link to a PDB formatted text file containing the coordinates of your protein and the matched protein superposed. There are many pieces of software available to view this data. Please see the Phyre2 FAQ for descriptions of these software packages, where you can get them, and their system requirements. |
Along the bottom of a row is a button to display matches to further members of this cluster. This is described below. |
Cluster members |
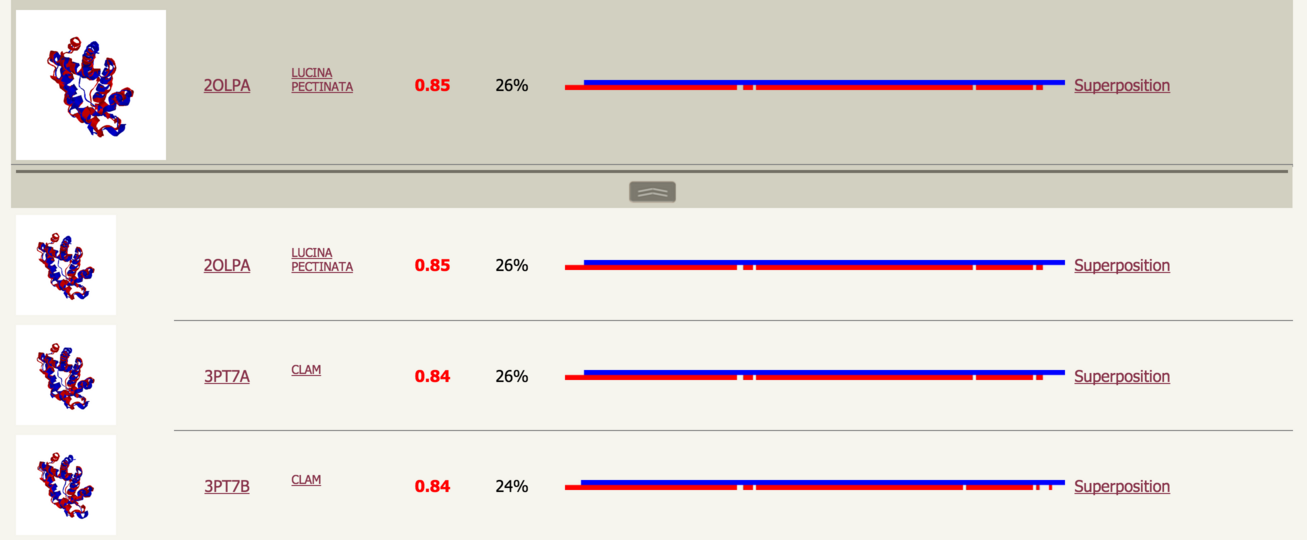 |
Clicking the button will reveal further matches to proteins in the cluster represented by the medoid. Intepretation of these results is just as with the medoids. |
TM-score |
| The TM-score was devised by Zhang and Skolnick in 2004 and is one of the de facto standards in use by the modelling community to measure the similarity between two protein structures. For details, please the Wikipedia entry. Intuitively, the TMscore can be understood as the fraction of amino acids in your structure that can be superposed on the matched structure within 4.0Å. |
Please cite
PhyreStorm: A web server for fast structural searches against the PDB.
Mezulis SM, Sternberg MJE & Kelley LA
Journal of Molecular Biology,
Volume 428, Issue 4, 22 February 2016, Pages 702–708
doi:10.1016/j.jmb.2015.10.017