|
| Help |
|
| Protein Function Detective | ||
PFD uses several methods of function prediction, using sequence and structure, to predict Gene Ontology (GO) terms for your protein. The information from these various methods constitutes the input to a machine learning algorithm called a Support Vector Machine (SVM). Using a benchmark of diverse proteins with known GO annotations, the SVM has been trained to discriminate between true and false positive annotations based on the outputs of the set of algorithms used in PFD. On our benchmark set of 125 proteins, PFD achieves 70% precision and 80% recall of GO terms.
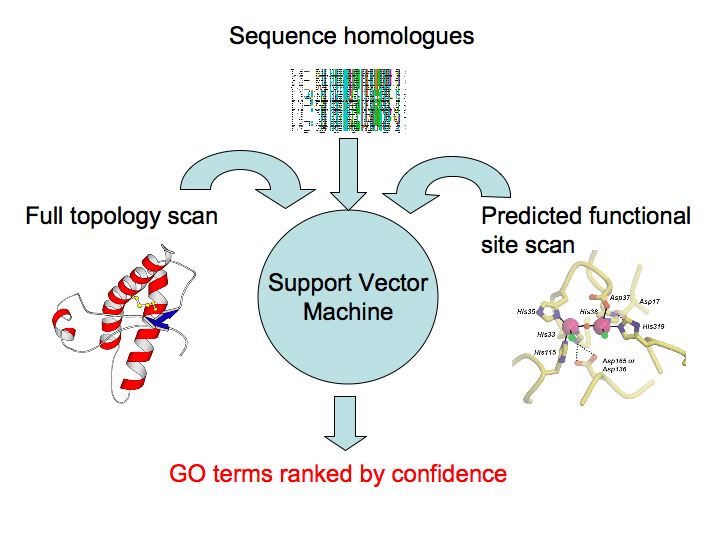
Full Topology SearchA full structural scan of your protein structure is made against the Structural Classification of Proteins (SCOP) database using a modified version of BLAST [1]. This converts your structure into a a string using a structural alphabet. This string is searched using the conventional BLAST algorithm against a database of pre-compiled structural sequences. Confident hits to known structures are stored, together with their associated GO terms. Functional Site PredictionPSI-Blast [2] is used to search for homologues of your protein sequence. These homologues are realigned using MUSCLE [3]. This alignment is used to perform a functional residue prediction using the Jenson-Shannon Divergence [4]. This is an information-theory approach to determine relative residue conservation. Such conservation is related to the functional importance of residues. Pocket/cleft DetectionDeep pockets or clefts in a protein are frequently associated with biological function. We detect such pockets by calculating the convex hull of a protein structure and its Delauney triangulation [5]. By calculating the conservation score of each pocket it is possible to rank the pockets for predicted functional relevance as well as the residues within each pocket. Functional Site SearchGiven candidate functional site residues, one may scan such sites against structures of known function using a fast geometric hashing technique [6]. This permits one to detect similar 3D patterns of residues, irrespective of connectivity, within other structures and rank potential matches by the quality of superposition and similarity of composition. Sequence HomologyUsing weak similarity to functionally annotated protein sequences in conjunction with the statistical relationships between the rate of co-occurrence of different biological functions has been shown to enhance functional annotation in the absence of clear homology [7]. Support Vector MachineSVMs are machine learning devices that allow one to quickly and accurately solve discrimination (and other) problems in the face of normally intractable high dimensional non-linear search spaces. We combine the confidence values, frequency of hits and background frequencies of GO terms from the above methods into a feature vector. Having trained an SVM on a benchmark of known structures, this vector can be classified as a true or false annotation. The output of the SVM can then be recast as a probability by fitting a probability distribution to training data. This final probability is that presented in the summary of results from a PFD run. References1.C.-H. Tung, J.-W. Huang and J.-M. Yang "Kappa-alpha plot derived structural alphabet and BLOSUM-like substitution matrix for fast protein structure database search," Genome Biology, vol. 8, pp. R31.1~R31.16, 2007. 2. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. SF Altschul, TL Madden, AA Schaffer, J Zhang, Z Zhang, W Miller and DJ Lipman. Nucleic Acids Research, Vol 25, Issue 17 3389-3402. 1997 3. Edgar, Robert C. (2004), MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Research 32(5), 1792-97. 4. Capra J, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics (2007) 23:1875 5. H. Edelsbrunner and M.Facello and Jie Liang. (1998). On the definition and the construction of pockets in macromolecules. [http://dx.doi.org/10.1016/S0166-218X(98)00067-5]. Discrete and Applied Mathematics, 88, 83-102. 6. M. Moll and L.E. Kavraki. Matching of Structural Motifs Using Hashing on Residue Labels and Geometric Filtering for Protein Function Prediction. The Seventh Annual International Conference on Computational Systems Bioinformatics (CSB2008), Stanford, CA, 2008. 7. Hawkins, T., Luban, S. and Kihara, D. 2006. Enhanced Automated Function Prediction Using Distantly Related Sequences and Contextual Association by PFP. Protein Science 15: 1550-6. Lawrence Kelley
| |||||||
