| Home | About | Contact | Download |
Method |
|
SuSPect uses sequence-, structure- and systems biology-based features to predict the phenotypic effects of missense mutations. 77 features are used to train a support vector machine (SVM) to discriminate between disease-causing and neutral variants. In a blind test from VariBench, SuSPect achieved an AUC (area under ROC curve) of 0.89, balanced accuracy of 82% and a Matthews correlation coefficient of 0.65, a large improvement over other methods tested. 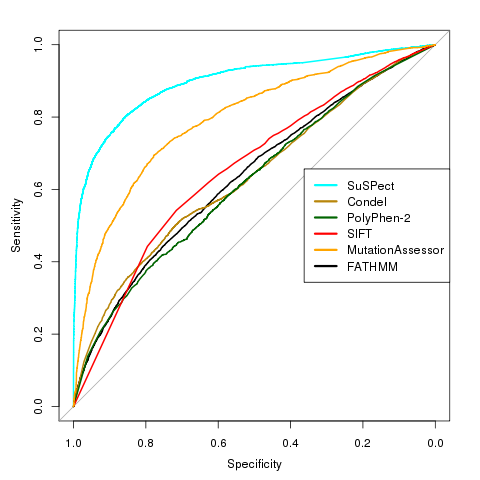 |
Usage |
Individual human variants - ExampleIndividual human variants can be uploaded and pre-calculated scores returned based on SuSPect-FS, a version of SuSPect using just 9 features which outperforms the full version. Where possible, these mutations can be mapped to known structures and homology models. Extra annotation is also available for each mutation, including known and predicted active sites, post-translational modifications and sequence conservation at that position. Annotation comes from Pfam, the Conserved Domain Database and UniProt. There are also predictions from TMHMM, NetPhos, NetNGlyc and NetOGlyc. SuSPect can currently accept mutations based on UniProt sequences (2013-03 release) or chromosomal co-ordinates, or as a VCF (variant call format) file, which will be mapped to UniProt sequences using ANNOVAR. For other sequences, try whole-protein mutation (see below). Variants should be either in UniProt or chromosomal co-ordinates: Whole protein mutation - ExampleSuSPect can accept a protein sequence, UniProt accession or structure file in PDB format. The structure file can be from an experiment or a homology model, for example from Phyre2. In the uploaded protein, all possible mutations are assessed and scores assigned. Scores for human proteins are pre-calculated using SuSPect-FS. For other proteins, a version of SuSPect has been developed that does not require PPI network features, and scores are calculated using this. If a structure has been provided, a PDB file will be made available to download with mean SuSPect scores in the temperature factor column. Individual positions of interest can be analysed by selecting a position and generating a PNG image or viewing the structure interactively with JSmol. |
Output |
|
SuSPect will produce a table of scores from 0-100, colour-coded according to predicted deleteriousness (blue=neutral, red=disease-causing). A score of 50 is recommended as a cut-off between neutral and disease-causing variants, with extreme scores being more confident predictions. Where a variant is known to be associated with a disease, for example in the training data, this will be noted, as will the presence of the variant in dbSNP. Clicking on a score will give more information about this variant, for example its predicted solvent accessibility, degree of conservation at that position, etc. There are also annotations from UniProt, the Catalytic Site Atlas, ProtInDB and PISite, in addition to predicted active sites (Conserved Domain Database, CDD), binding sites (CDD), post-translational modifications (NetPhos, NetNGlyc and NetOGlyc) and transmembrane helices (TMHMM). This extra information allows greater interpretation beyond simply having a score. |
SuSPectP |
|
SuSPect provides scores predicting whether or not a variant is likely to be associated with disease. However, in most cases a user is interested in a particular disease, so many variants predicted to be damaging will not be relevant for the disease of interest. With this in mind, SuSPectP has been developed to associate variants with a particular disease. After a SuSPect job has finished running, a link to submit the variants to SuSPectP will be available. SuSPectP works by combining the SuSPect scores with disease-specific scores calculated using PRINCE. Using SuSPectP, the correct variant is ranked in the top 10 up to 50 times more often than using SuSPect alone. An example of SuSPectP output is available here. |
Downloads |
SuSPect PackagePre-calculated scores for the entire human proteome are available to download here. The download includes an SQLite database which can be queried by UniProt accession or dbSNP rsID. There is also a Perl script which can accept chromosomal co-ordinates in addition to UniProt and rsID.SVMsThe SuSPect-All and SuSPect-FS SVMs are provided, together with the training data. As SuSPect is updated, these will be updated, but older versions will remain available.Test resultsThe predictions for the VariBench test set from SuSPect-All, SuSPect-FS, Condel, FATHMM, MutationAssessor, PolyPhen-2 and SIFT are provided. |
Structural Bioinformatics Group, Imperial College London
If you've found SuSPect useful, please cite our paper:
Yates CM, Filippis I, Kelley LA & Sternberg MJE (2014) SuSPect: Enhanced prediction of single amino acid variant (SAV) phenotype using network features. Journal of Molecular Biology. In press. http://dx.doi.org/10.1016/j.jmb.2014.04.026