Fold recognition methods and links
Some links for methods of FOLD recognition:
- Some links for methods that run via the WWW:
- Methods where an executable or code is available:
- Other relevant links:
Even with no homologue of known 3D structure, it may be possible to find a suitable fold for you protein
among known 3D structures by way of fold recognition methods
3D structural similarities
Ab initio prediction of protein 3D structures is not possible at present, and a general solution
to the protein folding problem is not likely to be found in the near future. However, it has
long been recognised that proteins often adopt similar folds despite no significant sequence
or functional similarity and that nature is apparently restricted to a limited number of protein folds.
There are numerous protein structure classifications now available via the WWW:
- SCOP (MRC Cambridge)
- CATH (University College, London)
- FSSP (EBI, Cambridge)
- 3 Dee (EBI, Cambridge)
- HOMSTRAD (Biochemistry, Cambridge)
- VAST (NCBI, USA)
Thus for many proteins (~ 70%) there will be a suitable structure in the database from which to
build a 3D model. Unfortuantely, the lack of sequence similarity will mean that many of these go
undetected until after 3D structure determination.
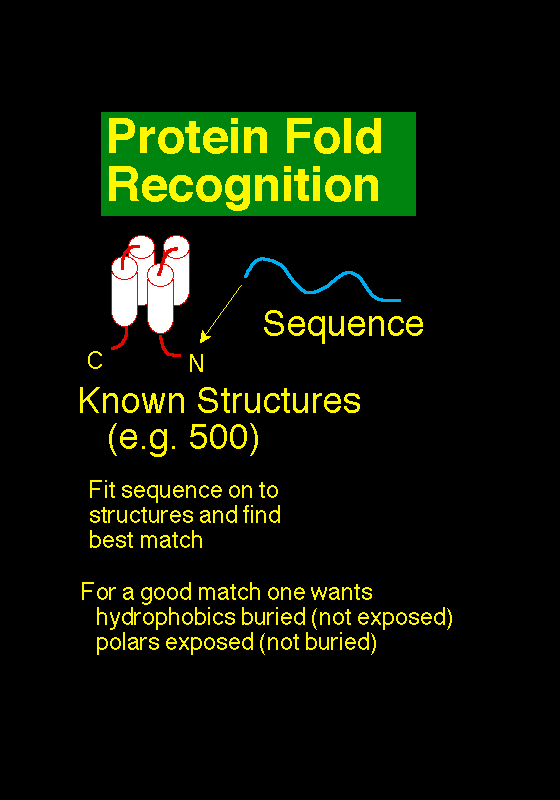
The goal of fold recognition
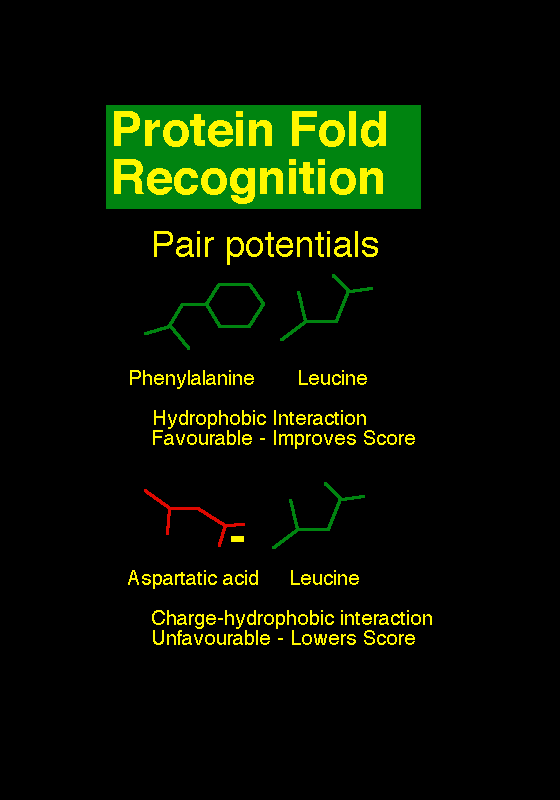
Methods of protein fold recognition attempt to detect similarities between protein 3D structure that
are not accompanied by any significant sequence similarity. There are many approaches, but the
unifying theme is to try and find folds that are compatable with a particular sequence. Unlike
sequence-only comparison, these methods take advantage of the extra information made available
by 3D structure information. In effect, the turn the protein folding problem on it's head: rather
than predicting how a sequence will fold, they predict how well a fold will fit a sequence.
Some papers on the subject:
- Reviews
- Wodak, S. J. & Rooman, M. J. (1993) Generating and testing protein folds, Current Opinion in Structural Biology, 3, 247-259.
- Jones, D. & Thornton, J. (1993) Protein fold recognition, Journal of Computer Aided Molecular Design, 7, 439-456.
- Bowie, J. U. & Eisenberg, D. (1993) Inverted protein structure prediction, Current Opinion in Structural Biology, 3, 437-444.
- Lemer C., Rooman, M. J. & Wodak, S. J. (1996), Protein Structure Prediction By Threading Methods: Evaluation Of Current Techniques, PROTEINS: Structure, Function and Genetics, 23, 337-355. (Assessment of techniques)
-
- Specific methods (these are now too numerous to mention, I just mention the earliest methods here, and some
that are available via the WWW)
- Ponder, J. W. & Richards, F. M. (1987), Tertiary templates for proteins: use of packing criteria in the enumeration of allowed sequence for dinfferent structural classes, Journal of Molecular Biology, 193, 775-791.
- M. J. Sippl, Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins, Journal of Molecular Biology, 213, 859-883. (PROFIT)
- Bowie, J. U., Luthy, R. & Eisenberg, D. (1991), A Method to Identify Protein Sequences That Fold into a Known Three-Dimensional Structure, Science, 253, 164-170.
- Jones, D.T., Taylor, W.R & Thornton, J.M (1992), A new approach to protein fold recognition, Nature,358, 86-89. (THREADER).
- Bryant, S. H. & Lawrence, C. E. (1993), An empirical energy function for threading a protein sequence through the folding motif, PROTEINS: Structure, Function and Genetics, 16, 92-112.
- Godzik, A., Kolinski, A. & Skolnick, J. (1992), Toplogy fingerprint approach to the inverse protein folding problem, Journal of Molecular Biology, 227, 227-238.
- Rost, B. (1995) TOPITS: Threading One-dimensional Predictions Into Three-dimensional Structures, The third international conference on Intelligent Systems for Molecular Biology (ISMB), 314-321. (TOPITS)
- Alexandrov, N. N., Nussinov, R. & Zmmer, R. M. (1995), Pacific Symposium on Biocomputing 1996 (Hunter, L. and Klein, T.E eds), 53-72. (123D)
- Have folds been predicted correctly? Yes. Here are some examples in the literature:
- Crawford, I. P., Niermann, T. & Kirschner, K. (1987), Predictions of secondary structure by evolutionary comparison: Application to the a
lpha subunit of tryptophan synthase, PROTEINS: Structure, Function and Genetics, 1, 118-129.
The structure was correctly predicted to adopt an alpha/beta barrel fold
- Bazan, J. F. (1990), Structural Design and Molecular Evolution of a Cytokine Receptor Superfamily,Proceedings of the National Academy of Science, 87, 6934-6938.
The structure was correctly predicted to adopt an Ig-type fold
- Gerloff, D. L., Chelvanayagam, G. & Benner, S. A. (1995), A predicted consensus structure for the protein-kinase c2 homology (c2h) domain, the repeating unit of synaptotagmin, PROTEINS: Structure, Function and Genetics, 22, 299-310.
The structure was correctly predicted to adopt a plastocyanin-type fold (though two alternative folds were given)
- Edwards, Y. J. K. & Perkins, S. J., (1995) The protein fold of the von Willebrand factor type A is predicted to be similar to the open twisted beta-sheet flanked by alpha-helices found in human ras-p21, 358, 283-286.
The structure was correctly predicted to adopt a ras-p21 type fold
The realities of fold recognition
Despite initially promising results, methods of fold recognition are not always accurate.
Guides to the accuracy of protein fold recognition can be found in the proceedings of the Critical
Assessment of Structure Predictions (CASP) conferences.
At the first meeting in 1994 (CASP1)
the methods were found to be about 50 % accurate at best
with respect to their ability to place a correct fold at the top of a ranked list. Though many methods failed
to detect the correct fold at the top of a ranked list, a correct fold was often found in the top 10 scoring folds.
Even when the methods were successful, alignments of sequence on to protein 3D structure were usually
incorrect, meaning that comparative modelling performed using such models would be inaccurate.
The CASP2 meeting held in December 1996, showed that many of the methods had improved, though it is difficult
to compare the results of the two assessments (i.e. CASP1 & CASP2) since very different criteria were used
to assess correct answers. It would be foolish and over-ambitious for me to present a detailed assessment of
the results here. However, and important thing to note, was that Murzin & Bateman managed to
attain near 100% success by the use of careful human insight, a knowledge of known structures, secondary
structure predictions and thoughts about the function of the target sequences. Their results strongly support
the arguments given below that human insight can be a powerful aid during fold recognition. A summary of the results from this meeting can be found in the PROTEINS issue dedicated to the meeting (PROTEINS, Suppl 1, 1997).
The CASP3 meeting was held in December 1998. It showed some progress
in the ability of fold recognition methods to detect correct protein folds
and in the quality of alignments obtained. A detailed summary of the
results will appear towards the end of 1999 in the PROTEINS supplement.
For my talk, I did a crude assessment of 5 methods of fold recognition. I took 12 proteins of known structure
(3 from each folding class) an ran each of the five methods using default parameters. I then
asked how often was a correct fold (not allowing trival sequence detectable folds) found
in the first rank, or in the top 10 scoring folds. I also asked how often the method
found the correct folding class in the first rank.
The results are summarised in here
in a PostScript file.
Perhaps the worst result from this study is shown below:
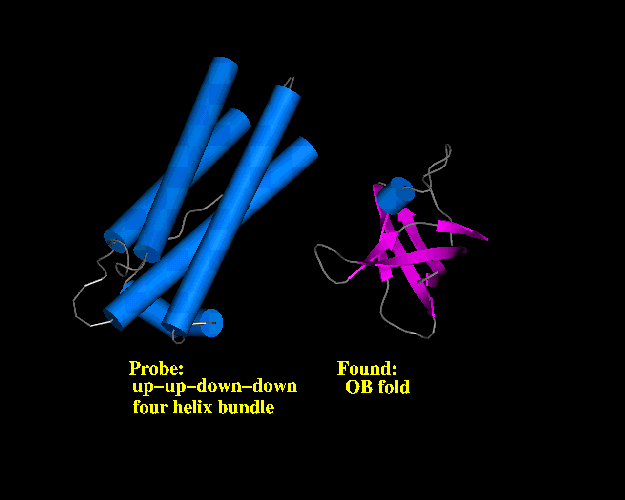
One method suggested that the sequence for the Probe (left)
(a four helix bundle) would best fit onto the structure shown on the right (an OB fold,
comprising a six stranded barrel).
The results suggest that one should use caution when using these methods. In spite of this,
the methods remain very useful.
A practical approach:
Although they are not 100 % accurate, the methods are still very useful. To use the methods I would suggest the following:
- Run as many methods as you can, and run each method on as many sequences (from your homologous protein family)
as you can. The methods almost always give somewhat different answers with the same sequences. I have also found that
a single method will often give different results for sets of homologous sequences, so I would also suggest running each
method on as many homologoues as possible. After all of these runs, one can build up a
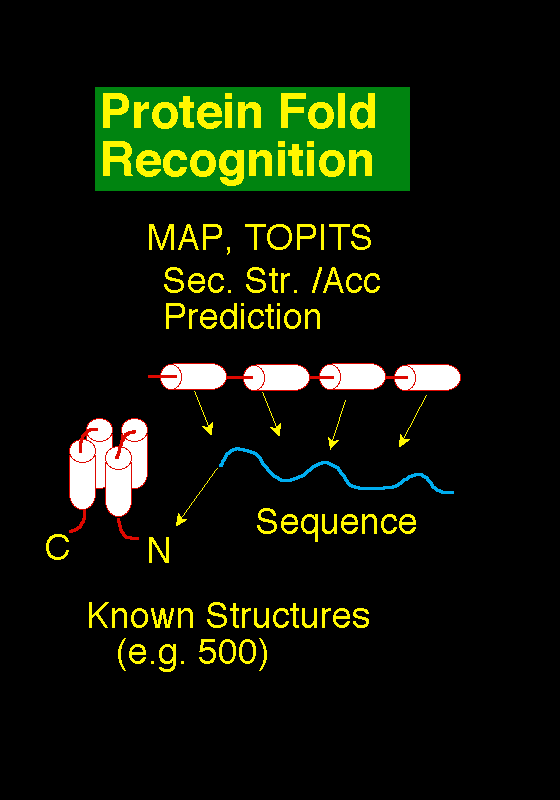
consensus picture of the likely fold in a manner similar to that used for secondary structure prediction above.
- Remember the expected accuracy of the methods, and don't use them as black-boxes. Remember that a correct
fold may not be at the top of the list, but that it is likely to be in the top 10 scoring folds.
- Think about the function of your protein, and look into the function of the proteins that have been found
by the various methods. If you see a functional similarity, then you may have detected a weak sequence homologue, or remote homologue.
At CASP2, as said above, Murzin & Bateman managed to obtain remarkably
accurate predictions by identification of remote homologues. Their
paper appeard in the PROTEINS supplement for the
CASP2 experiment:
Murzin AG, Bateman A (1997) Distant homology recognition using structural classification of proteins Proteins, Suppl 1, 105-112.
and provides some key insights into protein fold recognition using
humans rather than computers.
- Don't trust the alignments that are output by the programs. They can be used as a starting point, but the
best alignment of sequence on to tertiary structure is still likely to come from careful human intervention. One strategy for doing this is discussed in the next section
Fold recognition slides from my talk:
Slide 16
Slide 17
Slide 18
Slide 19
Slide 20
Slide 21
Slide 22
Slide 23
Slide 24
Slide 25
Slide 26
Slide 27
Next Analysis of folds and alignment of secondary structures.
Back to the Flowchart
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}