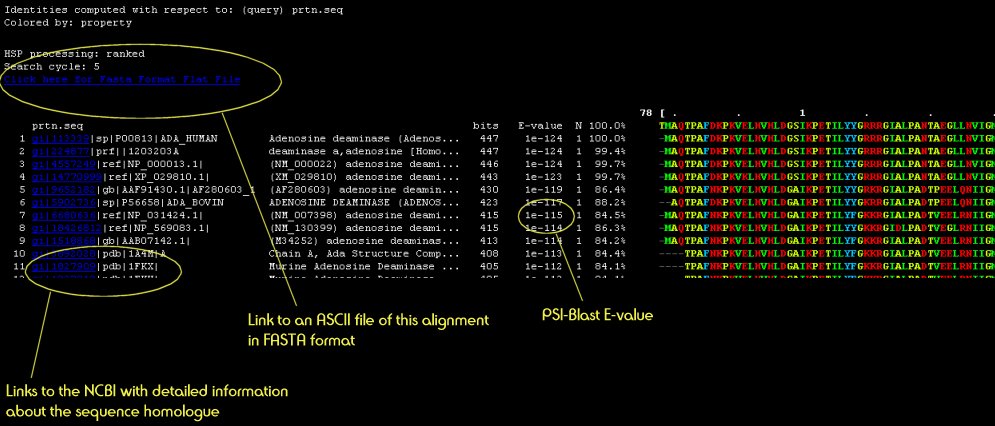
Your query sequence is scanned against an up-to-date sequence library to detect sequence homologues and to generate a sequence profile for your protein. On the results pages is a button labelled "View Multiple Sequence Alignment" Clicking this button opens a window looking something like this:
| The program PSI-Pred is used to generate a 3-state prediction (Helix/Strand/Coil) for your protein. This information is used later in the full structural search and is presented on the main results page for convenience. |  |
| Again, the PSI-Blast results are filtered and keyword information extracted. This is to provide some level of human-readable functional information about your protein. |

|
Fold Library
Each entry in the library is either a SCOP-defined domain, or a whole PDB chain. SCOP-defined domains are designated as d[pdbcode][chain][region]. In the case shown below, this is a SCOP-defined domain from PDB code 1cf7 for all of chain a.
In addition, whole PDB chains are added to the library when it is automatically updated. These codes begin with a 'c.

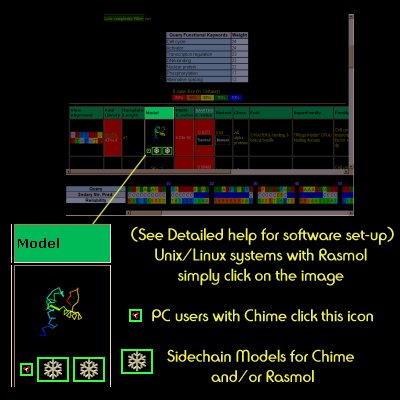
Model
The models generated by 3D-PSSM are simple mappings from the coordinates of the template structure and the query sequence residues aligned to them. If a confident hit is found, the model is further enhanced by the addition of sidechains using the SCWRL algorithm
How to view the models is dependent upon the computing platform you are using. On a PC or Mac platforms you should download the FREE Chime Plug-in from www.mdlchime.com and most things should work fine. On Linux or Unix you should download Rasmol and set it up as described in "Detailed Help"
E-value
The E-value is the measure of confidence in the prediction.

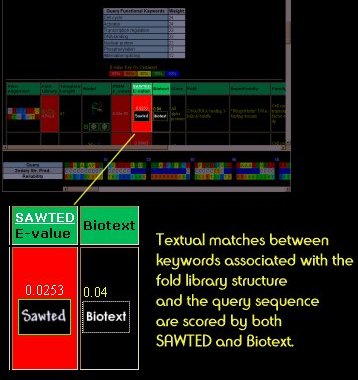
Text Matching
Two systems for matching functional keywords between your query protein sequence and template structures are used: SAWTED and Biotext. As with ordinary E-values, the closer this score is to zero, the better the textual match. Clicking on the Sawted or Biotext boxes will tell you the shared keywords.
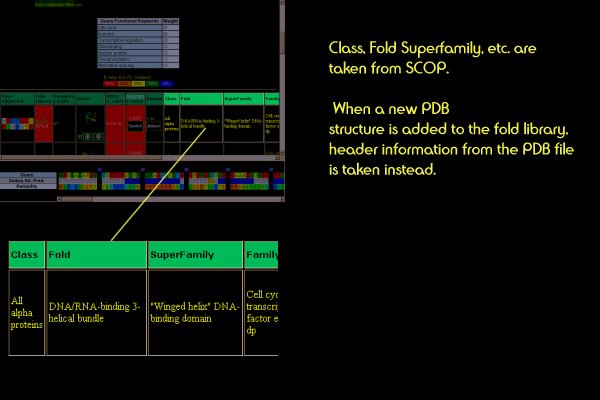
Structure/Function Classification
The classification of the template protein that best matches your query is shown.
3D-PSSM works by finding the best structural match to some region of your query sequence. Often this may mean 3D-PSSM finds a confident hit, but only to one portion of your query sequence. If this is the case, it is worth chopping out the remaining, unassigned region of your query and resubmitting this to the server.
In the near future some of this process will be automated, and the server will attempt to automatically detect multiple domains in your sequence.
This is a difficult question to answer. Issues of relevance are:
The E-values returned by 3D-PSSM are quite reliable, but occasionally false positives will appear. A much more common case is where 3D-PSSM has found an appropriate structural match, but the E-value is insignificant. This is where looking at other features such as the alignment can be important.
The text scores can sometimes indicate a correct hit lower down the table. As the text matching uses a quite different source of information to the sequence/structure matching, it can provide novel information. To some extent this is incorporated into the ranking already - confident text matching scores can move structures up the ranking. Looking at the keywords that are common between the query and template is always a good idea.
If the alignment is not too "gappy" and the secondary structure types align reasonably well, this is a sign of a decent alignment. The hydrophobic "core" residues of the template structure are colour-coded and numerically labelled in the alignment view. If these residues are not aligned to hydrophobic residues in the query, this can be a sign of trouble. Also, be wary of broken hydrophobic beta strands.
Finally, model quality. The models are annotated with the length and position of insertions and deletions. If a large deletion of the template structure is seen and the distance between the endpoints of the deletion cannot be bridged by neighbouring residues, this can also be a sign of trouble. However, often the choice of template may be correct, but the alignment may be wrong. This is a difficult call.
In summary, there are no hard and fast rules, but looking at all the sources of information together, including biological knowledge, is the best policy.